
Noticia completa disponible en:
LA OPS ACTUALIZA LA LISTA DE MEDICAMENTOS ESENCIALES PARA INCLUIR TRATAMIENTOS CLAVE CONTRA EL CÁNCER Y LA DIABETES
08 de septiembre 2025
La Organización Panamericana de la Salud (OMS) publica hoy ediciones actualizadas de sus listas modelo de medicamentos esenciales y medicamentos pediátricos esenciales, que incorporan nuevos fármacos para distintos tipos de cáncer y para la diabetes con afecciones asociadas, como la obesidad. También se han añadido tratamientos contra otras enfermedades, como la fibrosis quística, la psoriasis, la hemofilia y los trastornos hematológicos.
La 24ª Lista Modelo OMS de Medicamentos Esenciales y la 10ª Lista Modelo OMS de Medicamentos Pediátricos Esenciales incluyen los medicamentos que responden a las necesidades de salud prioritarias de las personas. Más de 150 países las han adoptado como referencia para adquirir y suministrar medicamentos en sus sistemas públicos de salud y para los sistemas de seguro y de reembolso.
USO DE PARACETAMOL DURANTE EL EMBARAZO
22 de septiembre 2025La FDA enfatiza que no se ha demostrado una relación causal, y que existen estudios con resultados contradictorios. No obstante, el riesgo potencial parece más relevante en casos de uso prolongado o crónico durante la gestación. A pesar de ello, la agencia reafirma que el tratamiento de la fiebre alta en embarazadas continúa siendo clínicamente justificado.
Como medida preventiva, la FDA ha iniciado el proceso de actualización del etiquetado de productos que contienen paracetamol y ha informado a los prescriptores en todo el país sobre esta nueva evidencia.
LA OMS ACTUALIZA LA LISTA DE MEDICAMENTOS ESENCIALES PARA INCLUIR TRATAMIENTOS CLAVE CONTRA EL CÁNCER Y LA DIABETES
08 de septiembre 2025La 24ª Lista Modelo OMS de Medicamentos Esenciales y la 10ª Lista Modelo OMS de Medicamentos Pediátricos Esenciales incluyen los medicamentos que responden a las necesidades de salud prioritarias de las personas. Más de 150 países las han adoptado como referencia para adquirir y suministrar medicamentos en sus sistemas públicos de salud y para los sistemas de seguro y de reembolso.
TERAPIAS FARMACOLÓGICAS PARA DIABETES TIPO 2
26 de agosto 2025La gama de tratamientos farmacológicos para la DT2 ha aumentado considerablemente, y los nuevos agentes han demostrado no solo eficacia hipoglucemiante, sino también una reducción de las complicaciones cardiometabólicas a largo plazo.
Esta revisión analiza los agentes farmacológicos más recientes para el tratamiento de la DT2 y la evidencia sobre sus beneficios cardiometabólicos. Se destacan las consideraciones clave para su uso según las características del paciente y el contexto clínico. Además, se analizan las terapias farmacológicas emergentes dirigidas a la patogénesis subyacente de la DT2, lo que subraya los avances continuos en el cuidado de la diabetes.
DIEZ PAÍSES DE LAS AMÉRICAS REPORTAN BROTES DE SARAMPIÓN EN 2025
15 de agosto 2025Los países más afectados son Canadá, México y Estados Unidos, donde también se concentran la mayoría de las muertes. Los brotes se relacionan con dos genotipos del virus. La OPS insiste en que el sarampión es altamente contagioso pero prevenible con dos dosis de la vacuna, por lo que urge reforzar la vacunación rutinaria y campañas en comunidades de riesgo.
La OPS brinda apoyo técnico en la región mediante vigilancia, diagnóstico, campañas de vacunación y envío de expertos a países afectados como México, Argentina y Bolivia.
NUEVA INYECCIÓN PARA FACILITAR LA PREVENCIÓN DE LA INFECCIÓN POR VIH EN LA UE Y EN TODO EL MUNDO
25 julio 2025La infección por VIH-1 reviste gran importancia para la salud pública. Según la OMS, en 2024 se estima que 1,3 millones de personas contrajeron la infección por VIH en todo el mundo, incluidas 160.000 nuevas infecciones en la región europea y 650.000 en África, la región más afectada por el VIH.

INNOVACIÓN EN SALUD PÚBLICA: BOLIVIA CAPACITA EN TRATAMIENTOS LOCALES EFECTIVOS PARA LA LEISHMANIASIS CUTÁNEA
09 de julio 2025Esta iniciativa surge como respuesta a los desafíos que plantea esta enfermedad, considerada un problema de salud pública significativo en Bolivia, especialmente en regiones endémicas donde el acceso a los tratamientos sistémicos convencionales es limitado. Para superar estas barreras, el programa se enfocó en fortalecer las capacidades técnicas del personal de salud en el uso de alternativas terapéuticas locales, como la termoterapia y la aplicación intralesional de antimoniato de meglumina, siguiendo las directrices de la OPS/OMS.
El objetivo principal de la capacitación fue mejorar las competencias del personal médico y de enfermería en el diagnóstico y manejo clínico de la leishmaniasis cutánea no complicada. Los nuevos métodos permitirán ofrecer una atención más centrada en el paciente, que es a la vez más rápida, de menor toxicidad y con costos reducidos. Además, su aplicabilidad es ideal para zonas rurales o de difícil acceso.
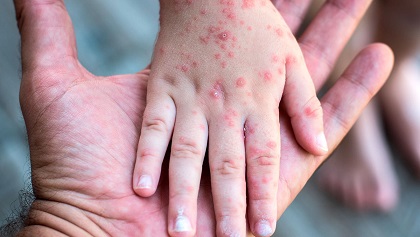
SARAMPIÓN 2025
25 junio 20251 Desde 2024, todas las regiones de la Organización Mundial de la Salud (OMS) han notificado un aumento del número de casos de sarampión, con 395.521 casos de sarampión confirmados por laboratorio notificados en 2024 y 16.147 notificados durante los 2 primeros meses de 2025.
2 Los pacientes de más de la mitad de los casos notificados fueron hospitalizados, por lo que el número real es probablemente mucho mayor.
3 Esta revisión abarca las presentaciones clínicas y las complicaciones del sarampión, las recomendaciones actuales y los antecedentes epidemiológicos del sarampión. También aborda los debates actuales sobre la inmunización y el tratamiento del sarampión y presenta información sobre los orígenes de las distintas vacunas antisarampionosas y actualizaciones sobre las pruebas diagnósticas del sarampión y los genotipos moleculares.
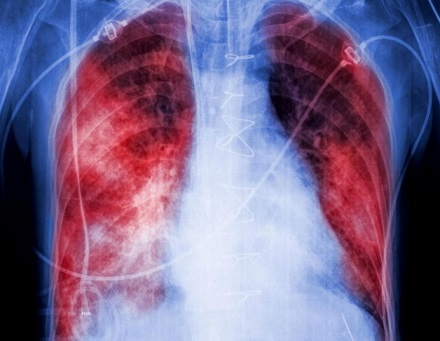
NERANDOMILAST EN PACIENTES CON FIBROSIS PULMONAR PROGRESIVA
18 de junio 2025
Métodos: En un ensayo clínico doble ciego de fase 3, se asignó aleatoriamente a pacientes con fibrosis pulmonar progresiva, en una proporción 1:1:1, a recibir nerandomilast en dosis de 18 mg dos veces al día, nerandomilast en dosis de 9 mg dos veces al día o placebo, con estratificación según el tratamiento de base (nintedanib frente a ninguno) y el patrón fibrótico en la tomografía computarizada de alta resolución (patrón similar a la neumonía intersticial habitual frente a otros patrones). El criterio de valoración principal fue el cambio absoluto con respecto al valor basal en la capacidad vital forzada (CVF), medida en mililitros, en la semana 52.
Resultados: Un total de 1176 pacientes recibieron al menos una dosis de nerandomilast o placebo, de los cuales el 43,5 % recibía tratamiento de base con nintedanib al inicio. El cambio medio ajustado en la CVF en la semana 52 fue de -98,6 ml (intervalo de confianza [IC] del 95 %: -123,7 a -73,4) en el grupo de nerandomilast 18 mg, de -84,6 ml (IC del 95 %: -109,6 a -59,7) en el grupo de nerandomilast 9 mg y de -165,8 ml (IC del 95 %: -190,5 a -141,0) en el grupo placebo. La diferencia ajustada entre el grupo de nerandomilast 18 mg y el grupo placebo fue de 67,2 ml (IC del 95 %, 31,9 a 102,5; p < 0,001), y la diferencia ajustada entre el grupo de nerandomilast 9 mg y el grupo placebo fue de 81,1 ml (IC del 95 %, 46,0 a 116,3; p < 0,001). El evento adverso más frecuente fue la diarrea, notificada en el 36,6 % de los pacientes del grupo de nerandomilast 18 mg, el 29,5 % del grupo de nerandomilast 9 mg y el 24,7 % del grupo placebo. Se presentaron eventos adversos graves en porcentajes similares de pacientes en los grupos del ensayo.
Conclusiones: En pacientes con fibrosis pulmonar progresiva, el tratamiento con nerandomilast produjo una disminución menor de la CVF que el placebo durante un período de 52 semanas. (Financiado por Boehringer Ingelheim; número de FIBRONEER-ILD en ClinicalTrials.gov, NCT05321082.)
CAMBIOS EN EL USO DE AZITROMICINA
23 mayo 2025
La azitromicina se utiliza desde hace décadas para tratar una amplia gama de enfermedades infecciosas, tanto en niños como en adultos. Está incluida en la lista de medicamentos esenciales de la Organización Mundial de la Salud (OMS), lo que pone de relieve su importancia para la salud pública.
Sin embargo, la azitromicina también está clasificada por la OMS como un antibiótico que conlleva un mayor riesgo de resistencia a los antimicrobianos y está incluida en la categoría de vigilancia de la OMS (clasificación AWaRe). Los datos muestran que la resistencia antimicrobiana contra este antibiótico ha aumentado en los últimos años.
Los medicamentos de la categoría Watch de la OMS deben priorizarse como objetivos clave de uso prudente y seguimiento. Sin embargo, los datos de consumo indican un aumento del uso de medicamentos con azitromicina en los últimos años. Un reciente estudio encargado por la EMA, realizado por DARWIN EU, mostró un amplio uso de este antibiótico en toda la UE, tanto en adultos como en niños.
Para promover un uso más racional de este antibiótico basado en las pruebas actuales y preservar su eficacia, el CHMP reevaluó los beneficios y riesgos de los medicamentos con azitromicina administrados por vía oral o en infusión (goteo) en una vena para los distintos usos autorizados

MEDIDAS PARA MINIMIZAR EL RIESGO DE PENSAMIENTOS SUICIDAS CON MEDICAMENTOS QUE CONTIENEN FINASTERIDA Y DUTASTERIDA
08 de mayo 2025
Los comprimidos de finasterida de 1 mg y el aerosol cutáneo de finasterida se utilizan para tratar la alopecia androgenética temprana, mientras que los comprimidos de finasterida de 5 mg y las cápsulas de dutasterida de 0,5 mg se utilizan para tratar la hiperplasia prostática benigna.
Después de una revisión de la Unión Europea de la información disponible sobre los medicamentos que contienen finasterida y dutasterida, el comité de seguridad de la EMA (PRAC) ha confirmado como efecto secundario de los comprimidos de finasterida de 1 y 5 mg la idea suicida (pensamientos suicidas).
La mayor parte de los casos de ideas suicidas fueron reportados en personas que habían tomado comprimidos de finasterida de 1 mg, los cuales son empleados para tratar la alopecia androgenética. La ficha informativa de los medicamentos que contienen finasterida ya incorpora una advertencia relacionada con alteraciones en el estado de ánimo, como la depresión, el ánimo deprimido y pensamientos suicidas. Los pacientes que presenten alteraciones en su estado de ánimo deben acudir a un médico y, en caso de estar utilizando finasterida de 1 mg, también es recomendable interrumpir el tratamiento.

NUEVO TRATAMIENTO CONTRA LA DISTROFIA MUSCULAR DE DUCHENNE
25 abril 2025La DMD es una enfermedad genética rara causada por la ausencia de distrofina, una proteína que ayuda a fortalecer las fibras musculares y a protegerlas de lesiones durante la contracción muscular. Esto causa que los músculos se debiliten gradualmente y pierdan su función, lo que culmina en la muerte. No existe cura para la DMD y el tratamiento se centra en la terapia con corticosteroides, la prevención de contracturas y el cuidado médico del corazón y la función respiratoria.
El principio activo de Duvyzat es el givinostat, un inhibidor de la histona desacetilasa (HDAC) que modula la actividad descontrolada de la HDAC en los músculos distróficos. Inhibir la actividad de la HDAC puede ayudar a reducir la inflamación y la cicatrización y el engrosamiento tisular.
Los eventos más frecuentes en pacientes tratados con givinostat fueron diarrea, dolor abdominal, trombocitopenia, vómitos, hipertrigliceridemia y fiebre.
Aún no se dispone de los datos a largo plazo de Duvyzat, la EMA solo respalda una licencia condicional, un procedimiento que permite el acceso temprano a medicamentos prometedores cuando existe una necesidad médica no cubierta.
Como condición, el laboratorio responsable deberá realizar estudios adicionales para confirmar la eficacia y seguridad del tratamiento, incluyendo ensayos controlados y un seguimiento basado en un registro de pacientes.

LA AEMPS REFUERZA LAS RECOMENDACIONES PARA MINIMIZAR EL RIESGO DE SOBREDOSIFICACIÓN ACCIDENTAL DE RISPERIDONA SOLUCIÓN ORAL EN PACIENTES PEDIÁTRICOS
11 abril 2025La Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) informa sobre la notificación de casos de sobredosis accidental con risperidona solución oral en población pediátrica debido a errores en la interpretación de las jeringas o pipetas que acompañan al medicamento. La mayoría de los casos notificados fueron graves y se relacionaron con errores en los decimales, con la consiguiente administración de 10 veces la dosis pautada.
La risperidona está indicada, en población pediátrica, en el tratamiento sintomático a corto plazo de la agresión persistente en los trastornos de la conducta en niños a partir de 5 años y en adolescentes con trastorno del desarrollo intelectual en los que la gravedad de la agresión u otros comportamientos perturbadores requieran tratamiento farmacológico.
Se recomienda a los profesionales sanitarios que proporcionen instrucciones claras a los pacientes y cuidadores sobre el uso correcto de los dosificadores indicando a los pacientes que presten atención al medir una dosis pequeña.
Los pacientes no deben utilizar un dosificador diferente al facilitado con el medicamento.
Después de la revisión del PRAC, se van a reforzar los mensajes sobre el manejo de los dispositivos para pacientes y cuidadores en el prospecto

COMITÉ DE LA EMA RECOMIENDA AMPLIAR LA INDICACIÓN DE MEDICAMENTOS PARA TRATAR OTRAS MUTACIONES RARAS DE FIBROSIS QUÍSTICA
31 marzo 2025La fibrosis quística es una enfermedad hereditaria con graves efectos en los pulmones, el sistema digestivo y otros órganos. Se acompaña de una alta mortalidad prematura. La fibrosis quística puede estar causada por diversas mutaciones en el gen de una proteína denominada «regulador de la conductancia transmembrana de la fibrosis quística». La proteína CFTR regula la producción de moco en los pulmones y de jugos digestivos en el intestino. Algunas mutaciones afectan el funcionamiento de la proteína, lo que provoca una excesiva densidad de la mucosidad y los jugos digestivos, lo que provoca obstrucciones, inflamación, mayor riesgo de infecciones pulmonares y problemas digestivos y de crecimiento. Otras mutaciones, llamadas mutaciones de clase I, impiden la producción de la proteína CFTR.
Kaftrio y Kalydeco se utilizan en combinación para tratar a pacientes cuya fibrosis quística se debe a al menos una mutación en el gen CFTR que causa una producción anormal o insuficiente de la proteína CFTR. Estos medicamentos actúan conjuntamente para mejorar la función de estas proteínas CFTR anormales, atacando así la causa subyacente de la enfermedad. Sus acciones combinadas reducen la densidad de la mucosidad pulmonar y los jugos digestivos, lo que ayuda a aliviar los síntomas de la enfermedad.

EL COMITÉ DE EVALUACIÓN DE RIESGOS (PRAC) EVALUARÁ EL RIESGO DE NEUROPATÍA ÓPTICA ISQUÉMICA ANTERIOR NO ARTERÍTICA (NAION)
25 febrero 2025La semaglutida, un agonista del receptor GLP-1, es el principio activo de ciertos medicamentos utilizados en el tratamiento de la diabetes y la obesidad (en concreto, Ozempic, Rybelsus y Wegovy).
El PRAC está evaluando si los pacientes tratados con semaglutida podrían presentar un mayor riesgo de desarrollar NAION. Este trastorno se produce por la reducción del flujo sanguíneo al nervio óptico, con posible daño al nervio, lo que puede provocar pérdida de visión en el ojo afectado. Los pacientes con diabetes tipo 2 podrían presentar un riesgo inherentemente mayor de desarrollar esta afección. El PRAC revisará ahora todos los datos disponibles sobre la NAION con semaglutida, incluyendo datos de ensayos clínicos, vigilancia poscomercialización, estudios sobre el mecanismo de acción y la literatura médica (incluidos los resultados de los estudios observacionales).

LAS AMÉRICAS PODRÁN ACCEDER A LA VACUNA HPV9 DESDE MEDIADOS DE 2025 A TRAVÉS DEL FONDO ROTATORIO DE LA OPS
05 febrero 2025La vacuna VPH9-valente estará disponible para toda la población desde los 9 hasta los 26 años, según los esquemas de cada país. La OPS busca poner fin a más de 30 enfermedades en el 2030, entre ellas el cáncer cervicouterino vacunando al 90% de las niñas antes de los 15 años. Además, el Fondo Rotatorio facilita la adquisición de vacunas mediante negociaciones colectivas entre los países participantes.
NUEVA COMBINACIÓN DE MEDICAMENTOS PARA TRATAR INFECCIONES POR GUSANOS PARÁSITOS
31 enero 2025
La ivermectina/albendazol tiene ventajas en programas de salud pública, como reducir los costos y mejorar la adherencia al tratamiento. En ensayos clínicos, demostró ser más eficaz que el albendazol solo para tratar ciertos parásitos. Es importante para países donde estas enfermedades son endémicas, y su evaluación se realizó bajo el procedimiento EU-M4all, en colaboración con la OMS y países objetivo. Los efectos secundarios comunes incluyen dolor de cabeza, dolor abdominal y enzimas hepáticas elevadas.
Este medicamento facilitará el tratamiento de millones de personas en zonas de riesgo, contribuyendo a la lucha contra enfermedades tropicales desatendidas.
ADVERTENCIAS DE SEGURIDAD MÁS DESTACADAS SOBRE LOS EFECTOS NEUROPSIQUIÁTRICOS DEL MONTELUKAST
16 enero 2025Se está agregando información de seguridad adicional a todos los productos de montelukast para reforzar y resaltar las advertencias existentes sobre eventos neuropsiquiátricos graves.
Estos incluyen cambios de comportamiento, depresión y pensamientos y comportamientos suicidas. La información de seguridad incluye: una nueva advertencia en recuadro, orientación adicional para prescriptores y pacientes sobre el manejo de eventos neuropsiquiátricos graves. Esta actualización de seguridad sigue a una investigación de seguridad de la TGA realizada en 2024 después de que los reguladores internacionales reforzaran las advertencias sobre eventos neuropsiquiátricos para los productos de montelukast.
Noticia completa disponible en:
MEDICAMENTOS HUMANOS EN 2024
16 enero 2025En 2024, la EMA recomendó la autorización de 114 medicamentos, de los cuales 46 contenían principios activos nuevos para la Unión Europea. Destacaron varios medicamentos innovadores, la EMA recomendó el primer medicamento para tratar el Alzheimer en etapas iniciales, una nueva forma de adrenalina sin aguja para reacciones alérgicas, y el primer tratamiento para tumores relacionados con la enfermedad de von Hippel-Lindau, así como antibióticos para el tratamiento de infecciones graves. También se recomendaron nuevas vacunas, incluyendo una para la enfermedad de Chikungunya y una vacuna de ARNm contra el virus respiratorio sincitial (VSR), además de ampliar el uso de una vacuna mpox para adolescentes.
El cáncer fue el área terapéutica más abordada, con 28 productos oncológicos recomendados, y también se incluyeron 28 recomendaciones para nuevos productos biosimilares para tratar diversas enfermedades. Los biosimilares son importantes para facilitar el acceso a tratamientos médicos vitales.
Tras la autorización de la Comisión Europea, la EMA y los Estados miembros de la UE supervisan la calidad y seguridad de los medicamentos, adoptando medidas regulatorias cuando sea necesario.

PRIMER TRATAMIENTO RECOMENDADO PARA ENFERMEDADES PULMONARES PROGRESIVAS POCO FRECUENTES EN NIÑOS Y ADOLESCENTES
13 diciembre 2024Ofev está autorizado en la Unión Europea para tratar estas enfermedades en adultos. El principio activo de Ofev, el nintedanib, actúa bloqueando enzimas llamadas tirosina quinasas, implicadas en la formación de tejido cicatricial. Este mecanismo ayuda a reducir la formación de cicatrices pulmonares.
El medicamento se presenta en cápsulas que se administran dos veces al día y contará con una dosis menor para uso pediátrico. Dado que estas afecciones afectan a menos de 5 niños por millón en la UE, la recomendación del Comité de Medicamentos Humanos (CHMP) de la EMA se basó en un pequeño ensayo clínico pediátrico y en datos extrapolados de estudios realizados en adultos. Solo se tratará con Ofev a niños cuyas afecciones progresan, y la decisión deberá ser tomada por un equipo multidisciplinario de especiales
LA AEMPS ACTUALIZA LA SITUACIÓN DE SUMINISTRO DE LOS MEDICAMENTOS ANÁLOGOS DEL GLP-1
09 diciembre 2024Los análogos del GLP-1 están aprobados para mejorar el control glucémico en adultos con diabetes mellitus tipo 2 (DM2) que no logran resultados adecuados con dieta y ejercicio. Estos medicamentos pueden utilizarse como monoterapia, cuando la metformina no es adecuada por intolerancia o contraindicación, o en combinación con otros tratamientos antidiabéticos. Además, algunos de estos medicamentos están autorizados para el control del peso en ciertas circunstancias.
Hasta ahora, Saxenda estaba autorizado para el control del peso, pero recientemente se aprobó nuevas presentaciones de Wegovy, cuya comercialización está prevista para mayo de 2024. En ambos casos, el tratamiento para el control del peso debe combinarse con una dieta equilibrada y un aumento en la actividad física.
Por otra parte, dos medicamentos del mismo grupo, Byetta y Lyxumia, que se consideraron alternativas, han dejado de comercializarse.
Pese a las medidas adoptadas para afrontar la alta demanda de estos fármacos en el tratamiento de la diabetes, los problemas de suministro, que comenzaron a finales de 2022, persisten, afectando a los pacientes. Por este motivo, la AEMPS enfatiza la importancia de ajustar las prescripciones para priorizar su uso en el control glucémico de pacientes con DM2, dado que las alternativas terapéuticas pueden ser más complejas.
COLCHICINA EN EL INFARTO DE MIOCARDIO: UN ENSAYO CLÍNICO ALEATORIZADO
17 noviembre 2024Métodos: En este ensayo multicéntrico con un diseño factorial de 2 por 2, asignamos aleatoriamente a pacientes que habían sufrido un infarto de miocardio para que recibieran colchicina o placebo y espironolactona o placebo. Los resultados del ensayo con colchicina se informan aquí. El resultado primario de eficacia fue una combinación de muerte por causas cardiovasculares, infarto de miocardio recurrente, accidente cerebrovascular o revascularización coronaria no planificada impulsada por isquemia, evaluada en un análisis del tiempo hasta el evento. Se midió la proteína C reactiva a los 3 meses en un subgrupo de pacientes y también se evaluó la seguridad.
Resultados: Se aleatorizó a un total de 7062 pacientes en 104 centros en 14 países; en el momento del análisis, se desconocía el estado vital de 45 pacientes (0,6 %), y es muy probable que esta información no se haya obtenido al azar. Se produjo un evento de resultado primario en 322 de 3528 pacientes (9,1%) en el grupo de colchicina y 327 de 3534 pacientes (9,3%) en el grupo de placebo durante un período de seguimiento medio de 3 años (cociente de riesgos instantáneos, 0,99; intervalo de confianza del 95% [IC], 0,85 a 1,16; P = 0,93). La incidencia de componentes individuales del resultado primario pareció ser similar en los dos grupos. La diferencia media de mínimos cuadrados en los niveles de proteína C reactiva entre el grupo de colchicina y el grupo de placebo a los 3 meses, ajustada según los valores iniciales, fue de -1,28 mg por litro (IC del 95%, -1,81 a -0,75). Se produjo diarrea en un porcentaje mayor de pacientes con colchicina que con placebo (10,2% frente a 6,6%; P<0,001), pero la incidencia de infecciones graves no difirió entre los grupos.
Conclusiones: Entre los pacientes que sufrieron un infarto de miocardio, el tratamiento con colchicina, cuando se inició poco después del infarto de miocardio y se continuó durante una mediana de 3 años, no redujo la incidencia del resultado primario compuesto (muerte por causas cardiovasculares, infarto de miocardio recurrente, accidente cerebrovascular o revascularización coronaria no planificada provocada por isquemia). (Financiado por los Institutos Canadienses de Investigación en Salud y otros; número CLEAR ClinicalTrials.gov, NCT03048825)
SE RECOMIENDA LEQEMBI PARA EL TRATAMIENTO DE LA ENFERMEDAD DE ALZHEIMER EN ETAPA TEMPRANA
14 noviembre 2024Los pacientes con una o ninguna copia de ApoE4 tienen menor riesgo de desarrollar anomalías en las imágenes relacionadas con el amiloide (ARIA), un efecto adverso grave de Leqembi que puede provocar hinchazón y posible sangrado en el cerebro.
El CHMP concluyó que, en la población específica revisada en este nuevo análisis, los beneficios de Leqembi en la ralentización de los síntomas de la enfermedad son mayores que sus riesgos.

Noticia completa disponible en:
TRATAMIENTOS MÁS CORTOS CON ANTIBIÓTICOS
04 noviembre 2024En Canadá, se estima que en 2018 se produjeron 5400 muertes atribuibles a la RAM. Para 2050, el 40% de las infecciones serán resistentes a los antimicrobianos de primera línea. El plan de acción pancanadiense sobre la RAM ha delineado acciones prioritarias para responder a esta amenaza en Canadá, incluida la gestión de antimicrobianos.
Los tratamientos más prolongados con antibióticos no previenen la RAM; aumentan la probabilidad de efectos adversos y de RAM.
Históricamente, la duración del tratamiento con antibióticos ha sido fija, y se ha ajustado arbitrariamente al número de días de la semana sin que se haya demostrado o tenido en cuenta la mejoría clínica. Un error común es creer que los pacientes deben “terminar el tratamiento” incluso si se sienten mejor, para evitar la resistencia. Sin embargo, los resultados de una revisión general de ensayos controlados aleatorizados (ECA) encontraron que cada día adicional de uso de antibióticos se asociaba con un aumento de la resistencia a los antibióticos y un 4 % más de probabilidades de efectos adversos de los antibióticos, como exantema, diarrea y superinfecciones.
La mayoría de las infecciones agudas adquiridas en la comunidad se pueden tratar con 5 a 7 días de antibióticos.
Más de 130 RCTs en 22 enfermedades infecciosas han demostrado que los ciclos más cortos de antibióticos son equivalentes, o no inferiores, a los ciclos más largos. Entre estos estudios se encuentran los que muestran que un ciclo de 5 a 6 días equivale a 10 días para la celulitis, 3 a 5 días equivale a 5 a 14 días para la neumonía en niños y adultos, 5 días equivale a 7 días para las exacerbaciones agudas de la enfermedad pulmonar obstructiva crónica y 7 días equivale a 10 a 14 días para la pielonefritis en adultos.